Gibbs Free Energy
- 相变平衡应用:两相平衡(如固 – 液、液 – 气平衡)时,相变的吉布斯自由能变化ΔtrsG=0(平衡状态dG=0);结合G=H−TS,可得:ΔtrsG=ΔtrsH−TtrsΔtrsS=0⟹ΔtrsS=TtrsΔtrsH其中,ΔtrsH为Enthalpy of Transition,Ttrs为Transition Temperature
- Trouton’s Rule:多数有机液体的Entropy of Vaporization ΔvapS约为85 – 88 $\text{J}\cdot\text{K}^{-1}\cdot\text{mol}^{-1}$,可用于估算未知液体的ΔvapH
大多数 non-associative liquids,比如苯、汽油,分子间不形成氢键,变成气体时的混乱度变化(\(\Delta_{\text{vap}} S\))几乎一样,大概在\(85 \sim 88 \, \text{J·mol}^{-1}·\text{K}^{-1}\)之间;因此只要知道液体的沸点,就能估算它变成气体需要的能量(汽化焓)
Gibbs free energy的differential form
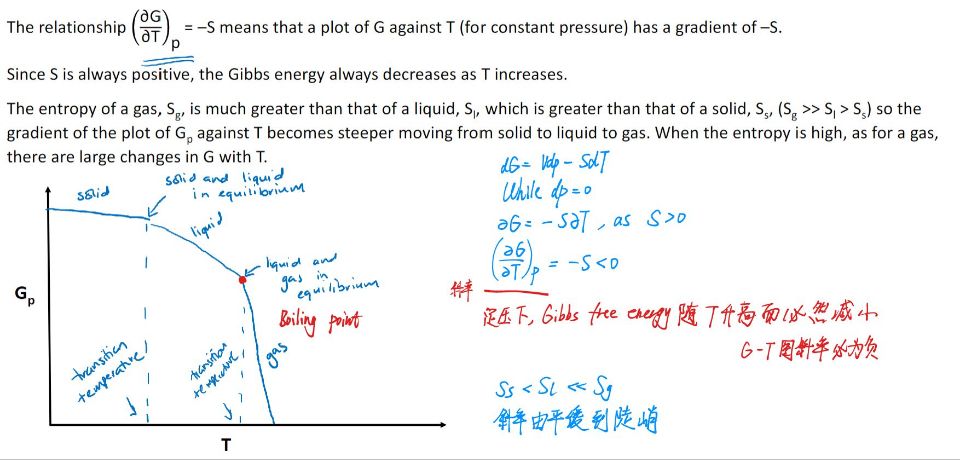
Variation of $G$ with $T$
$$(\frac{\partial G}{\partial T})p=−S$$
Variation of $G$ with $p$
$$(\frac{\partial G}{\partial p})T=V$$

不同相态的Molar Gibbs Free Energy
以恒温的条件为前提,即:
$$(\frac{\partial G}{\partial p})T=V$$
Condensed Phases:solid / liquid
condensed phases压缩性小,可假设体积\(V_m\)不随 $p$ 变化,积分得:$$G_m = G_m^\ominus + V_m \Delta p$$
其中$G_m^\ominus$为standard molar Gibbs free energy,\(p^\ominus=1\ \text{bar}\)),\(\Delta p = p – p^\ominus\)

Gas Phase
假设为ideal gas,\(V_m = RT/p\),积分得:$$G_m = G_m^\ominus + RT \ln\left(\frac{p}{p^\ominus}\right)$$
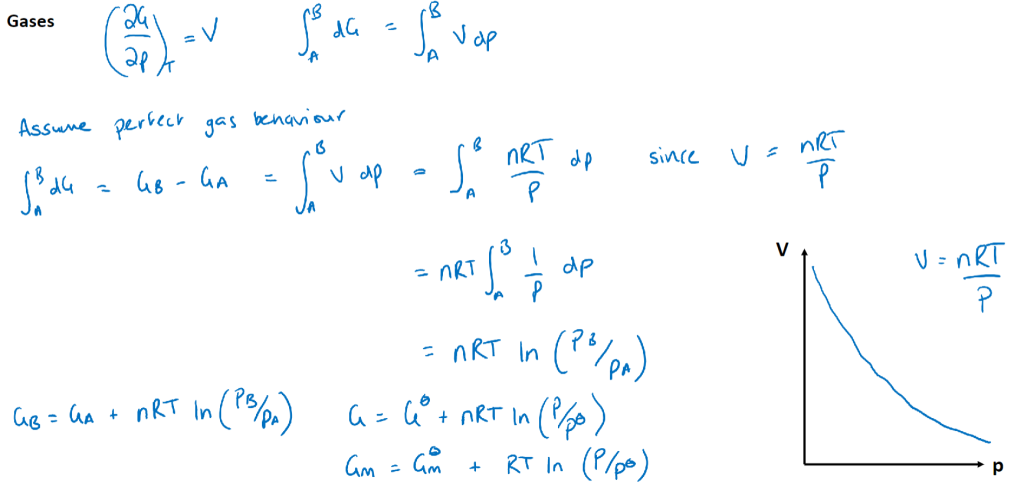
Phase transitions and equilibria
Definition
- Phase
物质中化学组成和物理状态均匀的部分.
- Vapour Pressure
condensed phase(固/液)与gas phase平衡时,气相产生的压强
气相压强在恒温下为定值
当vapour pressure等于外压时,液体开始沸腾,此时温度为boiling point $T_b$
- Phase Diagram $p-T$
以$p$为纵轴,$T$为横轴的图形,反应不同$p-T$条件下最稳定的相
Phase Rule
$$F=C-P+2$$
相律是为了计算$P$,其它量都会给出.
对于纯物质,Number of Components $C=1$
Number of Components是系统中Chemically Independent Species,即不能通过其它species的化学反应或浓度关系推导得到的species数量.
$$\boxed{C = S – R – R’}$$
- $S$:系统中所有物种的总数(如 “水 + 冰 + 水蒸气” 系统中,$S=1$,仅 $H_2O$)
- $R$:系统中 “独立的化学平衡反应数”(即不能由其他反应推导得到的化学反应)
- $R’$:系统中 “独立的浓度约束条件数”(如电荷平衡、浓度比例约束等)
示例:苯(A)+ 甲苯(B)的溶液 – 蒸气平衡
- 步骤 1:(S=2)(苯、甲苯,两相中物种相同)
- 步骤 2:无化学反应,(R=0)
- 步骤 3:无浓度约束(苯和甲苯比例可任意),$R’=0$
组分数 $C = 2 – 0 – 0 = 2$
相律 $F=2-P+2=4-P$,如单相时 F=3,可独立变 $ T$、$p$、浓度
相稳定性判断
$$G=H-TS$$
Gibbs Free Energy越小越稳定.
- 低温时:$TS$项影响小,Enthalpy占主导;固体 $G$ 最小,最稳定
- 高温时:$TS$项(负贡献)增大,Entropy主导;由于$S_{\text{固体}} < S_{\text{液体}} \ll S_{\text{气体}}$,气体 $G$ 最小,最稳定
- 高压时:$G=U+pV-TS$中,$pV$项主导,体积小(密度大)的相$pV$更小,$G $更低;例如:冰的$V_m$大于水(冰密度小),高压下水比冰稳定
Clapeyron Equation & Clausius-Clapeyron Equation
Proof of Clapeyron Equation
A, B两相平衡,则有$G_A=G_B$并且$dG_A=dG_B$;
由Gibbs Free Energy的differential表达式$dG=VdP-SdT$,代入得:
$$V_A dp – S_A dT = V_B dp – S_B dT$$
结合相变过程中熵的变化和体积的变化:
$$
\begin{cases}
&\Delta_{trs}S = S_B – S_A \\
&\Delta_{trs}V = V_B – V_A
\end{cases}
$$
最终得:
$$\frac{dp}{dT} = \frac{\Delta_{trs}S}{\Delta_{trs}V}$$
再结合相变平衡的$\Delta_{trs}S = \frac{\Delta_{trs}H}{T_{trs}}$,可改写为:
$$\frac{dp}{dT} = \frac{\Delta_{trs}H}{T_{trs}\Delta_{trs}V}$$
Properties of Clapeyron Equation
- 适用于所有类型的相变
- 物理含义:$\frac{dp}{dT}$是相边界的斜率,反映维持两相平衡时,$T$变化所需的$p$的变化率.
Clausius-Clapeyron Equation
推导(针对液 – 气相变)
对液 – 气相变(汽化),文档引入两个合理近似:
- 液体体积远小于气体体积:\(V_{\text{液体}} \ll V_{\text{气体}} \implies \Delta_{vap}V \approx V_{\text{气体}}\);
- 气相近似为理想气体:\(V_{\text{气体}} = \frac{RT}{p}\)(摩尔体积)
把$\Delta_{\text{vap}} V_m \approx \frac{RT}{p}$和 Molar Enthalpy of Vaporization $\Delta_{\text{trs}} H_m = \Delta_{\text{vap}} H_m$代入克拉佩龙方程:
$$\frac{dp}{dT} = \frac{\Delta_{\text{vap}} H_m}{T \cdot \frac{RT}{p}}$$
整理后得到:
$$\frac{dp}{dT} = \frac{p \Delta_{vap}H_m}{RT^2}$$
积分形式
假设\(\Delta_{vap}H_m\)不随 T 变化,对\(\frac{dp}{p} = \frac{\Delta_{vap}H_m}{R} \cdot \frac{dT}{T^2}\)从\((T_A, p_A)\)到\((T_B, p_B)\)积分,得:
\(\ln\left( \frac{p_B}{p_A} \right) = -\frac{\Delta_{vap}H_m}{R} \left( \frac{1}{T_B} – \frac{1}{T_A} \right)\)
适用场景
- 仅适用于有气相参与的相变(液 – 气、固 – 气,即汽化、升华)
- 升华过程可类比,将\(\Delta_{vap}H_m\)替换为摩尔升华焓(Molar Enthalpy of Sublimation, \(\Delta_{sub}H_m\)),公式形式不变
Chemical Potential $\mu$
chemical potential可以理解为物质在系统中的 “逃逸倾向” 或 “转移能力”
物质从\(\mu\)高的区域自发向化学势\(\mu\)低的相或系统流动,直到各相 / 系统的化学势相等(达到平衡)
多相平衡时,同一组分在各相中的\(\mu\)相等,如冰 – 水平衡时$\mu_{冰} = \mu_{水}$
Single-Component System
单组分系统中,$G$ 与物质的量 $n$ 成正比($G$ 是 Extensive property),定义Molar Gibbs Energy $G_m$为化学势:
$$\mu = G_m = \frac{G}{n}$$
对于single-component system,由于没有其它物质的影响,此时 $G$ 的微分式(含组成变化)为:
$$dG = Vdp – SdT + \mu dn$$
类比 Molar Volume
摩尔体积和化学势的定义,都是通过 “消除物质的量的影响”,将extensive property转化为intensive property
Multi-Component System
多组分系统有两种或更多物质(如盐水、空气),此时每个组分的化学势需考虑其他物质的影响,定义为偏摩尔吉布斯自由能.
partial molar Gibbs free energy
多组分系统(如 $A$、$B$ 混合物)中,$G$ 依赖 $p$、$T$ 及各组分的物质的量($n_A, n_B,\cdots$),定义组分 $j$ 的化学势为:
$$\mu_j = \left( \frac{\partial G}{\partial n_j} \right)_{T, p, n_{i \neq j}}$$
$T, p, n_{i \neq j}$表示 “固定温度、压强,且其他组分的物质的量不变”;
物理意义
向系统中加入 1 mol 组分 $j$(其他条件不变)引起的 $G$ 变化,也称为Partial Molar Gibbs Energy.
Ideal Solution的chemical potential
理想溶液中,分子间作用力均匀,可通过 Mole Fraction \(x_i\) 描述浓度对化学势的影响:
- Solvent ,组分 A 的chemical potential
遵循Raoult’s Law:
$$\mu_A = \mu_A^* + RT \ln x_A$$
\(\mu_A^*\):纯溶剂(\(x_A = 1\))的chemical potential;\(x_A\):溶剂的Mole fraction(\(0 < x_A < 1\));\(R = 8.314 \, \text{J·mol}^{-1}·\text{K}^{-1}\);$T$:绝对温度(K)
- Solute,组分 B 的chemical potential
稀溶液中遵循Henry’s Law,有两种表达形式:
- 以摩尔分数\(x_B\)表示:
$$\mu_B = \mu_B^\circ + RT \ln x_B$$
2. 以物质的量浓度\(c_B\)表示(\(c^\circ = 1 \, \text{mol·dm}^{-3}\)为标准浓度):
$$\mu_B = \mu_B^{\circ, c} + RT \ln \frac{c_B}{c^\circ}$$
$mu_B^\circ$、$mu_B^{\circ, c}$为溶质的标准化学势,与浓度标度有关
| 系统类型 | 化学势公式 | 关键变量与说明 |
|---|---|---|
| 单组分系统 | \(\mu = G_m\) | \(G_m\):摩尔吉布斯自由能 |
| \(\left( \frac{\partial \mu}{\partial T} \right)_p = -S_m\) | \(S_m\):摩尔熵;温度升高,化学势下降 | |
| \(\left( \frac{\partial \mu}{\partial p} \right)_T = V_m\) | \(V_m\):摩尔体积;压力升高,化学势上升 | |
| 多组分理想溶剂 | \(\mu_A = \mu_A^* + RT \ln x_A\) | \(\mu_A^*\):纯溶剂化学势;\(x_A\):溶剂摩尔分数 |
| 多组分理想溶质 | \(\mu_B = \mu_B^\circ + RT \ln x_B\) 或 \(\mu_B = \mu_B^{\circ, c} + RT \ln \frac{c_B}{c^\circ}\) | \(x_B\):溶质摩尔分数;\(c_B\):溶质浓度;\(c^\circ = 1 \, \text{mol·dm}^{-3}\) |
| 实际溶液 | \(\mu_i = \mu_i^\circ + RT \ln a_i\)(\(a_i = \gamma_i x_i\)) | \(a_i\):活度;\(\gamma_i\):活度系数(\(\gamma_i = 1\)为理想) |
在纯物质方程$dG = Vdp – SdT$基础上,加入组成变化的贡献:$dG = Vdp – SdT + \sum_j \mu_j dn_j$;
推广到其他热力学势($U$,$ H$, $A$):
$$dU = TdS – pdV + \sum_j \mu_j dn_j$$
$$dH = TdS + Vdp + \sum_j \mu_j dn_j$$
$$dA = -pdV – SdT + \sum_j \mu_j dn_j$$
不同势的化学势定义等价,仅偏导条件不同,考试重点为 $dG$ 的形式.
类比:电动势
离子化学势:\(\mu_j = \mu_j^\ominus + RT \ln a_j\)
电化学核心关系:\(\Delta G = -nFE\)
可以把 “电化学中的化学势” 理解为化学势在‘有电子转移的系统’中的应用;本质逻辑没变,只是多了 “电子” 这个参与物质,物质(包括离子、电子)都随化学势流动
Gibbs-duhelme equation
Appendix
考试易错点总结
混淆凝聚相与气相的 Gm 表达式
- 错误:用气相公式\(G_m = G_m^\ominus + RT \ln(p/p^\ominus)\)计算液体或固体的 G 变化;
- 正确:凝聚相用\(G_m = G_m^\ominus + V_m \Delta p\)(\(V_m\)小,\(\Delta G_m\)通常很小),气相用理想气体公式(\(\Delta G_m\)随 p 变化显著)。
相律中组分数$C$判断错误
- 错误:将混合物的 “物种数” 当作组分数(如 NaCl 水溶液,物种数为 3(NaCl、Na⁺、Cl⁻),但 C=2—— 物种数 – 1 个电离反应 = 3-1=2);
- 正确:\(C = \text{物种数} – \text{独立化学反应数} – \text{浓度约束数}\)(纯物质\(C=1\),无反应和约束)。
Clapeyron与Clausius-Clapeyron的适用场景混淆
- 错误:用克劳修斯 – 克拉佩龙方程计算固 – 液相变(如冰 – 水平衡);
- 正确:
- 克拉佩龙方程:适用于所有相变(固 – 固、固 – 液、液 – 气);
- 克劳修斯 – 克拉佩龙方程:仅适用于有气相的相变(液 – 气、固 – 气),且需满足\(V_{\text{凝聚相}} \ll V_{\text{气相}}\)。
化学势定义条件遗漏
- 错误:将\(\mu_j\)写成\(\left( \frac{\partial G}{\partial n_j} \right)_T\)(忘记固定 p 和\(n_{i \neq j}\));
- 正确:\(\mu_j = \left( \frac{\partial G}{\partial n_j} \right)_{T, p, n_{i \neq j}}\)—— 必须固定温度、压强和其他组分的物质的量,否则无物理意义
理想气体混合时误用总压代替分压
- 错误:计算\(G_{\text{最终}}\)时用总压 p 代替分压\(p_j\)(如\(G_{\text{最终}} = n_A (\mu_A^\ominus + RT \ln(p/p^\ominus)) + n_B (\mu_B^\ominus + RT \ln(p/p^\ominus))\));
- 正确:混合物中各组分的化学势由分压决定,\(p_j = x_j p\),需代入\(p_j\)计算(如\(\ln(p_A/p^\ominus) = \ln(x_A p/p^\ominus)\))
混合熵(\(\Delta_{mix}S\))的符号错误
- 错误:认为\(\Delta_{mix}S < 0\)(误判\(\ln x_j\)的符号);
- 正确:\(x_j < 1\),\(\ln x_j < 0\),故\(\Delta_{mix}S = -nR \sum x_j \ln x_j > 0\)(熵增,符合自发过程的熵增原理)
0 条评论