- U9
- U10 The Laws of Thermodynamics and Chemical Reactions
- U11 Kinetics
- Transition Metal Chemistry

U9
9.2 The Bonding in Organic Molecules
Hybridisation
- 杂化类型的判断方法
$$\text{杂化数} = \text{连接的原子数(和键数无关)}+ \text{孤对电子数}$$
$$\text{含双键就是} sp^2 \text{杂化},\text{含三键就是} sp \text{杂化}$$
杂化数(即杂化类型)决定了空间构型.
所谓双键、三键,之所以被称为“不饱和键”,就是因为它们是由未参与杂化的p轨道形成的.

HOMO and LOMO orbitals
并非独立于成键轨道、反键轨道之外的 “特殊分子轨道类型”,而是对分子轨道电子填充状态的一种描述. 它们反映了成键轨道和反键轨道在电子填充中扮演了怎样的角色.

9.3 Functional groups and oxidation level
- 基本官能团
- Oxidation and Reduction
氧化反应:得氧失氢、失电子
还原反应:失氢得氧、得电子
Inductive properties
诱导效应是电负性原子极化分子中 σ 键(σ – bonds)的方式
基本方式就是比较基团的电负性.
- 具有$-I$ electron-withdrawing effects的基团
Strong:$\ce{X-(X=F,Cl,Br,I)}$,$\ce{-O}$,$\ce{NH3}$
Weak:$\ce{OH-}$,$\ce{NH2-}$
- 具有$+I$ electron-donat ing effects的基团
$\ce{CH3+}$,$\ce{Li+}$,$\ce{MgBr+}$
Resonance
轨道离域(Orbital delocalisation)使 π 键电子(π -bonding electrons)能够在分子中移动,而非局限于某两个原子之间
当分子存在共轭体系(如多个双键交替排列)时,π 轨道相互重叠,形成离域的大 π 轨道,电子不再定域于单个双键,而是在整个共轭区域内离域
Carbocation Stability
Mesomeric properties
https://byjus.com/jee/mesomeric-effect
中介效应源于通过共振共轭(resonance conjugation)在分子的 π 体系(π – system)内的电子密度移动(the movement of electron density)

The group which shows the $–M$ effect includes:
$\ce{–NO2, –CN, –COX, –SO3H, – CHO, –CONH2, –COR, –COOH, –COOR}$ etc.
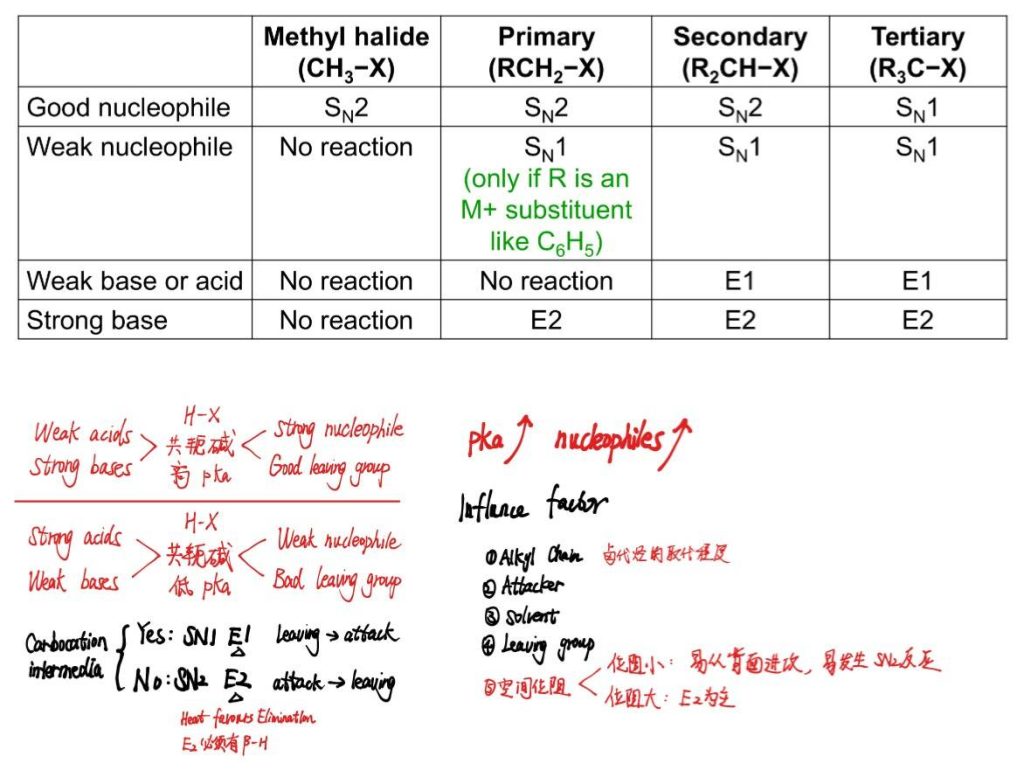
9.4 Nucleophilicity and Reaction Mechanisms
亲核反应机理:Nucleophile & Base
U10 The Laws of Thermodynamics and Chemical Reactions
Basic
Extensive & Intensive Properties
- extensive:与amount相关
质量、体积 - intensive:与amount无关
温度、压力
$$\frac{\text{extensive properties}}{\text{amout}}=\text{intensive properties}$$
$$V_m=\frac{V}{n}$$
$2^{nd}$ Law of Thermodynamics
- Spontaneous processes
- 热运动自发扩散不做功(Ideal mixing by diffusion)
- 气体向真空膨胀不做功(Joule Gas Expansion)
仅靠吸放热,无法预测物理或化学变化过程是否自发,必须有第二个判据.
- Entropy Change of the Surroundings ($\Delta S_{surr}$)
热力学第二定律即是针对反应自发性的结论.
The size of the entropy change depends on the temperature: $$\Delta S_{\text{surr}}=\frac{-\Delta H}{T}$$
- $2^{nd}$ Law of Thermodynamics
In a spontaneous process the total entropy of the Universe increases.
$$\Delta S_{\text{sys}} + \Delta S_{\text{surr}} \geq 0$$
\begin{aligned}
\Delta S_{\text{sys}} – \frac{\Delta H}{T} &\geq 0\\
T\Delta S_{\text{sys}} – \Delta H &\geq 0\\
0 &\geq \Delta H – T\Delta S_{\text{sys}}
\end{aligned}
- $\Delta G$ and Spontaneity
Gibbs Free Energy, $\Delta G$, is the maximum work that can be obtained from a change without expansion of the system.
$$\Delta G = \Delta H – T\Delta S$$
$$\Delta G=-RT\ln K$$
- $\Delta_r G<0$: The process is spontaneous.
- $\Delta_r G=0$: The process is at equilibrium.
- $\Delta_r G>0$: The reverse process is spontaneous.
A spontaneous process must have $\Delta G<0$ ,and $K>0$ (favouring products).
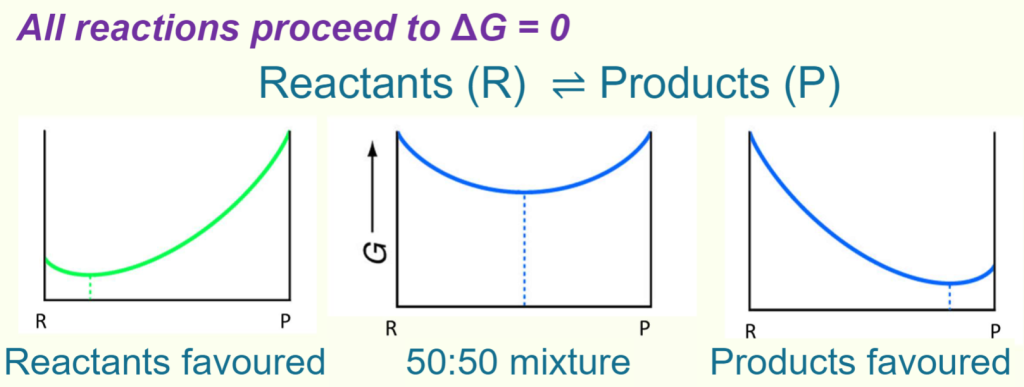
For an irreversible reaction, the equilibrium state is reached when all reactants are used up.
$Q$ – The Reaction Quotient
对于反应 $aA + bB \rightleftharpoons cC + dD$,其反应商 $Q_c$ 的表达式为:
$$Q_c = \frac{[C]^c [D]^d}{[A]^a [B]^b}$$
Gibbs Energy and Equilibria
$$G = G^\ominus + RT\ln Q$$
At equilibrium, $Q = K$
$\Delta G^\ominus = -RT\ln K$ and $\Delta G^\ominus = \Delta H^\ominus – T\Delta S^\ominus = 0$
$$\Delta H^\ominus – T\Delta S^\ominus = -RT\ln K$$
Heat, work and the First Law of Thermodynamics
The First Law of Thermodynamics
The First Law describes how the system interacts with the surroundings during a chemical change.

Internal Energy and Enthalpy
lecture10.2
- Work and Reversibility
在可逆过程中,系统能做 最大功(Maximum work);这是因为该过程中,体积膨胀导致的内能损失(internal energy loss)被最小化
10.6 Redox Reactions and Electrochemistry
Redox Reactions and Ions in Solution
U11 Kinetics
$Chemistry^3$在小绿鲸中大概408页左右.

Fundamental
反应物与生成物反应速率呈镜像关系,在同一时刻,反应物消耗速率$-\frac{d|A|}{dt}$与产物生成速率$-\frac{d|P|}{dt}$数值相等,符号相反(same magnitude,opposite sign)
Rate Coefficients
反应速率常数$k$是反应的固有属性,不同反应$k$值也不同:
$$A \xrightarrow{k_1} P_1,B \xrightarrow{k_2} P_2$$
$$k_1\ne k_2$$
Enzyme Kinetics
Enzyme对substrate浓度$[S]$有复杂依赖,因底物结合和产物解离的平衡影响速率:
$$\text{E} + \text{S} \rightleftharpoons \text{ES} \rightarrow EP \rightarrow E+P$$
Elementary Reactions
- Unimolecular
单分子反应遵循一级反应动力学,即$-\frac{d|A |}{dt}\propto [A]$
由此推导出速率方程:
$$-\frac{d|A |}{dt}= k_{uni}[A]$$
对方程两边积分:
$$\int\frac{1}{|A |}d[A]=-\int k_{uni}dt$$
$$ln[A]t=-k_{uni}t+c$$
当$t=0$时,$c=ln[A]_0$,得到:
$$ln[A]_t=-k_{uni}t+ln{A}_0$$
Anti-log:
$$[A]t=[A]_0\exp(-k_{uni}t)$$
- Bimolecular
对于双分子反应$A+B\rightarrow P,-\frac{d|A |}{dt}\propto [A][B]$
由此推导出速率方程:
$$-\frac{d|A |}{dt}= k_{bi}[A][B]$$
Measuring the Order of Reaction, and Parallel Reactions
Measuring the Order
当一种反应物大量过量时,可将原本的二级反应视作一级反应,这种情况称为 伪一级反应(pseudo – first – order reaction)
- Isolation Method
For the reaction: $aA + bB + cC \to P$
$$ \frac{-d[A]}{dt} = k[A]^a[B]^b[C]^c $$
If $[B]$ and $[C] \gg [A]$, the rate equation:
$$ \frac{-d[A]}{dt} = k[B]^b[C]^c $$
can be simplified by defining $k’ = k[B]^b[C]^c$, leading to:
$$ \frac{-d[A]}{dt} = k'[A]^a $$
This form is analogous to the simpler rate equations we encountered previously.
- Integral method

Solution
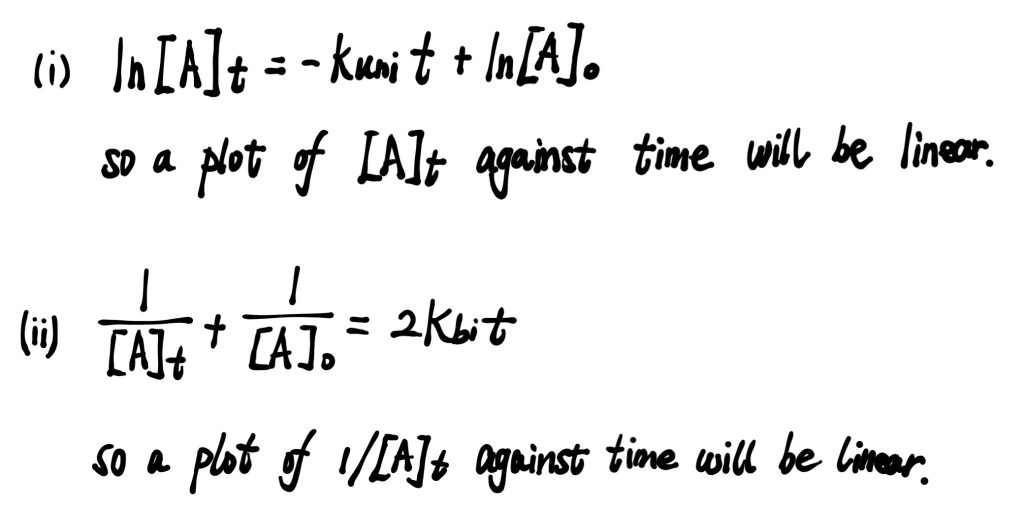
Where we determine the ‘shape’, or the relationship between $[A]$ and $t$.
- If a graph of $\ln{[A]}_t$ against $t$ is a straight line, then $a = 1$.
- If a graph of $\frac{1}{[A]_t}$ against $t$ is a straight line, then $a = 2$.
- Differential method

The differential of $[A]$ against $t$, is the gradient or rate.
We know that for elementary reactions: $\frac{d[A]}{dt} = k'[A]^a$, measuring gradients along one decay, and plot the gradients against $[A]$.
Or we take the initial gradient at $t = 0$, then repeat the experiment at different $[A]_0$ values.
11.3这部分,还需要复习一级反应和二级反应.
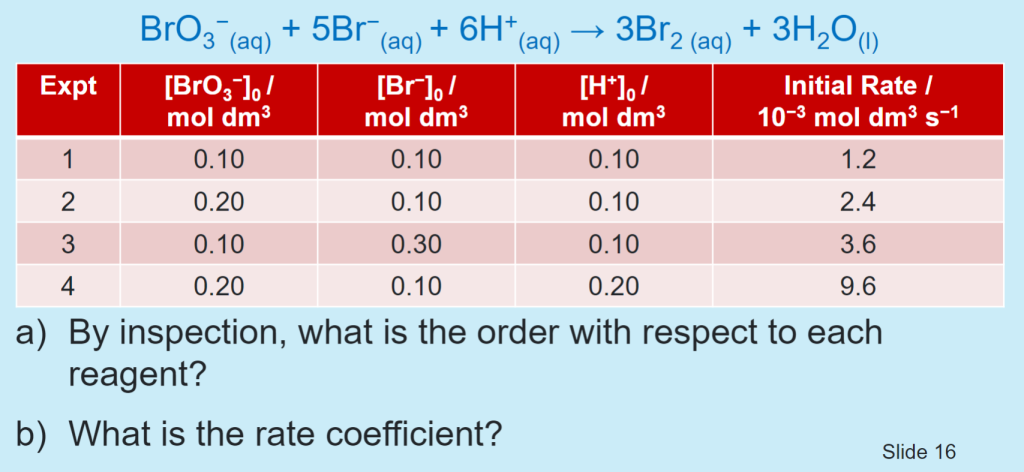
Solution


Transition Metal Chemistry
- 晶体场分裂
- 高低自旋
- 螯合效应
- CFAE
Crystal Field Theory
过渡金属的性质无法用主族元素的价键理论来解释(如orbital hybridisation),而crystal field theory即是考虑ligands如何影响金属$d$轨道的能量.
自由金属离子的 5 个 $d$ 轨道$d_{xy}, d_{xz}, d_{yz}, d_{z²}, d_{x²-y²}$能量完全相同,称为五重简并;配体的存在打破对称性,通过静电排斥(负点电荷配体与 $d$ 电子的排斥)导致轨道能量分裂,简并度降低.
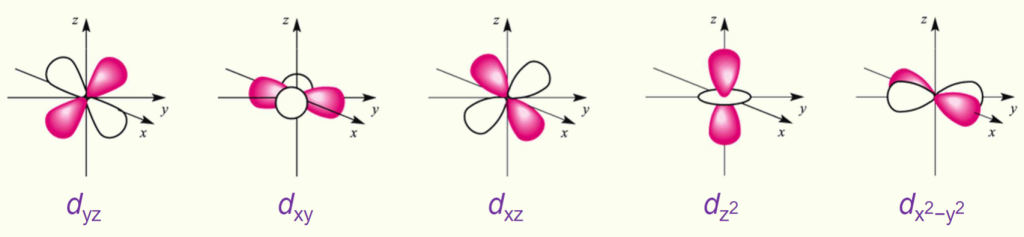
在晶体场理论中,将ligands视为negative point charges:
- 若配体是像$Cl^-$或$OH^-$这样的阴离子,其负电荷属性显而易见
- 若配体是像$H_2O$或$NH_3$这样的中性分子,负电荷则源于与孤对分子轨道(lone pair MO)相关的偶极(dipole):以$NH_3$为例,氮原子上的孤对电子形成偶极,一端为$δ-$(负电端),一端为$δ+$(正电端)
Octahedral Complexes
6 个配体沿 (x, y, z) 轴的正负 6 个方向分布(位于正八面体的 6 个顶点),这样就导致了$d$轨道的分裂:
- $d_{z^2}) 和 (d_{x^2−y^2}$ 轨道的波瓣(lobes,轨道的空间伸展方向)直接指向负点电荷,它们与配体(ligands)的排斥力大,因此能量更高
- $d_{yz}$、$d_{xy}$ 和 $d_{xz}$轨道的波瓣指向配体之间的空隙,电子受配体排斥力较小,所以能量更低
由此可知,在octahedral environment中,过渡金属$d$轨道分裂成两组:
- 能量较低的三个轨道$t_{2g}$
- 能量较高的两个轨道$e_g$
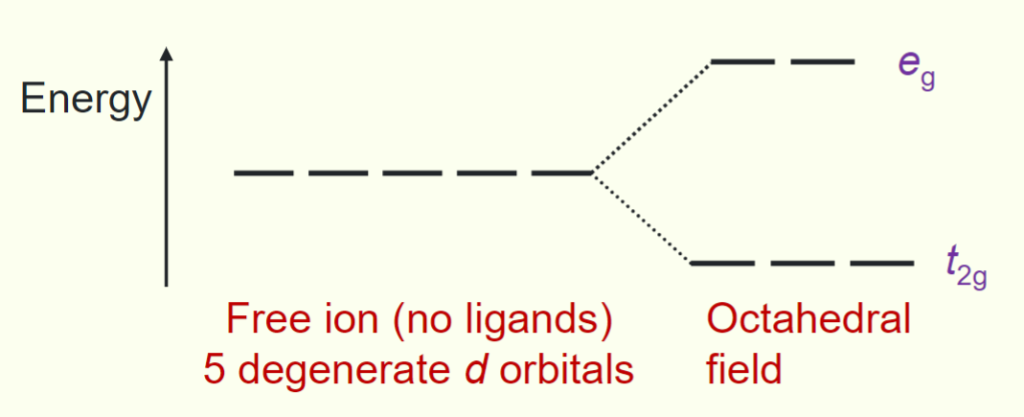
$\Delta_{oct}$ 表示晶体场分裂参数(crystal field splitting parameter)
- $t_{2g}$ 轨道的能量降低了 $-\frac{2}{5}\Delta_{oct}$($t_{2g}$ orbitals are lowered in energy by $-\frac{2}{5}\Delta_{oct}$);
- $e_g$ 轨道的能量升高了 $+\frac{3}{5}\Delta_{oct}$($e_g$ orbitals are raised in energy by $+\frac{3}{5}\Delta_{oct}$)
从配合物形成时整体 $d$ 轨道的能量变化来看,
$(2 \times \frac{3}{5}\Delta_{oct}) – (3 \times \frac{2}{5}\Delta_{oct}) = 0$
这表明虽然 $d$ 轨道发生了分裂,但总体能量保持不变,符合能量守恒原理(The overall $d$-orbital energy change from complex formation is $(2 \times \frac{3}{5}\Delta_{oct}) – (3 \times \frac{2}{5}\Delta_{oct}) = 0$)
Tetrahedral Crystal Field
Square planar
$d$轨道与ligand(negative point charges)的空间位置关系,决定了配合物各条$d$轨道的能量差异.
- (d_{z^2}) 轨道:在与负电荷(配体,可视为负点电荷)相同的平面内有一个小的 轨道波瓣(orbital lobe),但轨道的大部分远离 点电荷(point charges),与负电荷的静电排斥作用较弱,因此能量略低(slightly lower in energy)
- (d_{xz}) 和 (d_{yz}) 轨道:离负电荷最远(furthest away from the negative charges),与负电荷的静电排斥作用最弱,因此具有 最低的能量(lowest energy),且二者是简并的(degenerate),即能量相同
Electron Configurations
The Spectrochemical Series and the Jahn-Teller Effect
晶体场理论回顾(Recap – Crystal Field Theory)
晶体场理论(Crystal Field Theory, CFT)指出,过渡金属的d轨道能量会被配体(ligands)形成的负电荷场分裂,分裂方式取决于配体的几何构型(如八面体、四面体、平面正方形)及配体与d轨道的静电排斥作用。例如:
- 八面体场(Octahedral):d轨道分裂为能量较高的e_g轨道(d_{x²−y²}, d_{z²})和能量较低的t_{2g}轨道(d_{xy}, d_{xz}, d_{yz}),分裂能记为Δ_oct。
- 四面体场(Tetrahedral):分裂能Δ_tet约为Δ_oct的4/9,t₂轨道能量高于e轨道。
- 平面正方形场(Square planar):分裂能Δsp最大,d{x²−y²}轨道能量最高。
晶体场稳定能(Crystal Field Stabilisation Energy, CFSE)影响过渡金属配合物的结构、磁性、颜色和反应动力学。当分裂能Δ大于电子成对能(spin pairing energy, P)时,形成低自旋(Low spin, LS)配合物;反之形成高自旋(High spin, HS)配合物。
分裂能(Δ)的影响因素(Factors Controlling the Size of Δ)
- 金属氧化态(Metal oxidation state)
金属离子电荷越高,对配体的吸引力越强,Δ越大。例如:
- [Fe(H₂O)₆]²⁺的Δ_oct为9400 cm⁻¹,
- [Fe(H₂O)₆]³⁺的Δ_oct为13700 cm⁻¹。
高价态金属(如M(III))更易形成低自旋配合物。
- 过渡金属周期(Transition series of metal)
同一族中,第二、三过渡系金属的Δ大于第一过渡系(如Rh³⁺ > Co³⁺),因其d轨道扩展,电子成对能P较低,故第二、三过渡系配合物通常为低自旋。 - 同周期金属差异(Identity of the metal across a row)
同配体、同氧化态时,第一过渡系金属的Δ变化不规律,例如:
- [Ti(H₂O)₆]³⁺的Δ_oct为20300 cm⁻¹,
- [Fe(H₂O)₆]³⁺的Δ_oct为13700 cm⁻¹。
- 配合物几何构型(Geometry of the complex)
- 平面正方形(Square planar)Δ_sp最大,
- 八面体(Octahedral)Δ_oct居中,
- 四面体(Tetrahedral)Δ_tet最小(Δ_tet = 4/9 Δ_oct)。
- 配体类型(Identity of the ligand)
配体按分裂能从小到大排列形成光谱化学序列(Spectrochemical Series):
[
I^{-} < Br^{-} < Cl^{-} < F^{-} < H_{2}O < NH_{3} < en < bpy < phen < PR_{3} < CO \approx CN^{-}
]
弱场配体(Weak field ligands,如卤素离子)产生小Δ,强场配体(Strong field ligands,如CO、CN⁻)产生大Δ。晶体场理论无法解释该序列(如中性配体CO为强场),需结合分子轨道理论(MO theory)
配合物几何构型的预测(Understanding and Predicting Geometries)
- 四面体配合物(Tetrahedral Complexes)
弱场配体(如Cl⁻)、第一过渡系金属(如Ni²⁺, d⁸)或d⁷电子构型(如Co²⁺)倾向形成四面体,因Δ小,CFSE贡献低,如[NiCl₄]²⁻。 - 平面正方形配合物(Square Planar Complexes)
d⁸电子构型(如Ni²⁺、Rh(I))在强场配体(如CN⁻)或第二、三过渡系金属中易形成平面正方形,如[Ni(CN)₄]²⁻,此时CFSE最大且电子构型稳定(全满d轨道)。 - 八面体配合物(Octahedral Complexes)
多数d区金属形成八面体,因CFSE高,如[Ni(H₂O)₆]²⁺(Ni²⁺, d⁸)
Jahn-Teller效应(Jahn-Teller Effects)
理论核心:简并量子态会发生畸变以消除简并性
- Cu²⁺配合物(d⁹, t_{2g}⁶e_g³)
e_g轨道(d_{z²}, d_{x²−y²})上的电子分布不均导致简并,引发结构畸变: - 若d_{z²}轨道填充2个电子,d_{x²−y²}填充1个电子,配合物沿z轴拉长(elongation);
- 反之沿z轴压缩(compression)
畸变使两个电子进入低能轨道,一个进入高能轨道,整体能量降低 - 典例
[CuCl₆]⁴⁻、[CuBr₆]⁴⁻等八面体配合物常出现轴向拉长畸变。 - 适用电子构型
八面体中e_g轨道电子数为奇数的构型(如d⁴高自旋、d⁷低自旋、d⁹),四面体配合物中t₂轨道电子数不均的构型也可能发生Jahn-Teller畸变
关键结论(Lecture 12.4 Learning)
- 分裂能Δ的影响因素:金属氧化态、过渡系周期、配体类型及几何构型
- 光谱化学序列:实验确定的配体强弱顺序,强场配体导致大Δ,易形成低自旋配合物
3.几何构型与电子构型:
- d⁸金属离子(如Ni²⁺)在强场下呈平面正方形;
- 弱场或d⁷构型倾向四面体
- Jahn-Teller效应:消除简并态的结构畸变,常见于$e_g$轨道电子数不均的配合物(如Cu²⁺的d⁹构型)
通过上述因素,可预测配合物的磁性、结构及光谱性质,理解过渡金属化学的多样性
0 条评论