- General motion
- Solid State Physics
- Special Relativity
- Thermodynamics
- Relativity
General motion




Solid State Physics
Properties of electrons

Unit 2: Electronic Structure of Atoms
https://www.youtube.com/watch?v=EMq_QbyghMU
Particle in box是量子力学经典模型,涉及解决Time-Independent Schrodinger Equation:
$$-\frac{\hbar^2}{2m}\frac{\partial \varphi}{\partial x^2}=E\varphi$$
Unit 3: Bonding Between Atoms
Unit 4: Crystal
常识性的:

4.1 Motifs and Lattices
- 2D Bravais Lattices
总的来说,二维的布拉维格子可以归纳为4种:

在这四种shape以外的,都无法通过平移填满整个二维平面,因此无法作为valid unit cell.
- 3D Bravais Lattices

三维晶体必然由某种粒子按照以上七种bravais lattices形成motif,即unit cell.
当atom只分布在corner上时,这种unit cell,被称为是’primitive’的,而fcc,bcc此类不仅仅在corner上有原子的晶胞,就属于non-primitive. 原子不能随意分布,基于七种基本的primitive bravais lattices结构,一共有十四种bravais lattices.
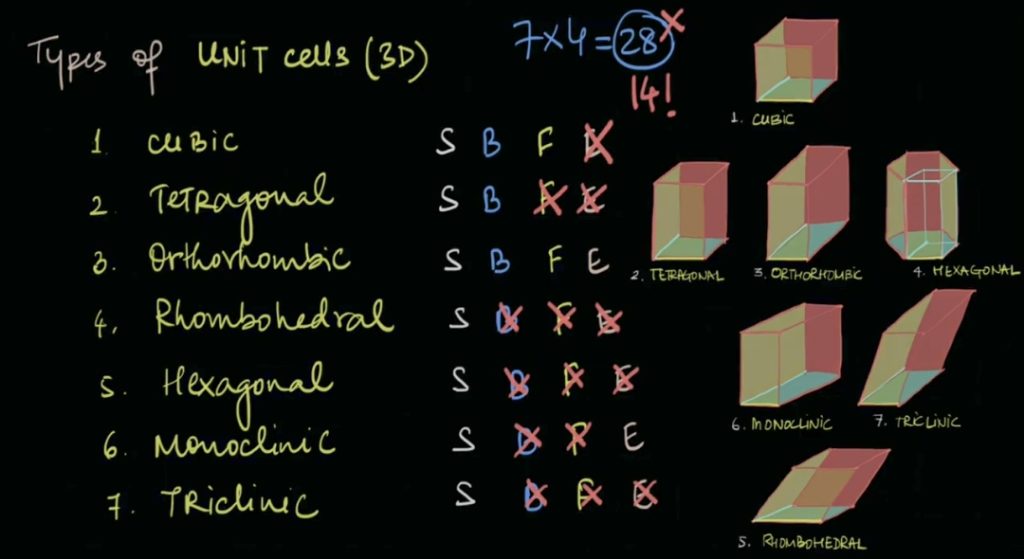
整理后如下,可以清晰看到衍生关系:

常见问题
如何判断一个二维点阵是否为布拉维晶格?
判断依据是点阵中所有点的环境是否相同.
若二维点阵中每一个点的周围环境(包括相邻点的数量、距离和排列方式等)都完全一致,即通过平移矢量 $\vec{T} = n\vec{a} + m\vec{b}$,(n, m 为整数)平移后,所有点的环境不变,则该二维点阵为布拉维晶格;若存在至少两个点的环境不同(如相邻点的数量、距离或排列有差异),则不是布拉维晶格.
一个晶格点上只能有一个原子吗?
一个晶格点上不一定只有一个原子.
一个晶格点(Lattice Point)是为了描述晶体周期性而引入的抽象几何点,并非实际原子,因此晶格点上可以没有原子,也可以有多个原子;例如,在一些复杂晶体结构中,一个晶格点可能对应一个基元(Motif),基元可包含多个原子.
有其他原子,不妨碍某种原子是布拉维晶格吗?
不妨碍.
布拉维晶格关注的是晶格点的周期性和每个晶格点环境的一致性,而非具体原子种类。即使存在其他原子,只要某种原子的排列方式满足:所有晶格点的环境相同(通过平移矢量 $\vec{T} = n\vec{a} + m\vec{b}$ 重复后,原子排列的相对关系不变),则这种原子的排列可构成布拉维晶格。例如,在 $\ce{NaCl}$ 结构中,虽然存在 $\ce{Na+}$ 和 $\ce{Cl-}$,但面心立方(FCC)的晶格点(顶点和面心)环境一致, $\ce{Na+}$ 或 $\ce{Cl-}$ 的排列均可基于此晶格描述,其他原子的存在不影响晶格本身的布拉维性质。
综上,晶格点与原子数量无必然限制关系,其他原子的存在也不影响某种原子的排列构成布拉维晶格,关键在于晶格点的周期性和环境一致性.
Planes in Lattices
Unit 5: Diffraction
Reference:https://www.youtube.com/watch?v=4JjhiyXcPl8
5.1 衍射的基本原理 – Huygens-Fresnel principle
Diffraction is the bending of a wave due to interference with an object.
光遇到障碍物时,我们观察到它会发生弯曲,我们用Huygens principle解释这个现象
空间中到处都有光的ether particles,无时无刻不在振荡,在wavefront上的每一个ether particle都会产生它们的次级波;换言之,wavefront上的每个点都充当一个新的source,产生一个新的spherical wave(球面波,即secondary wave),而所有点的所有次级波可以找到一个共同的envelope line,这就是新的wavefront:
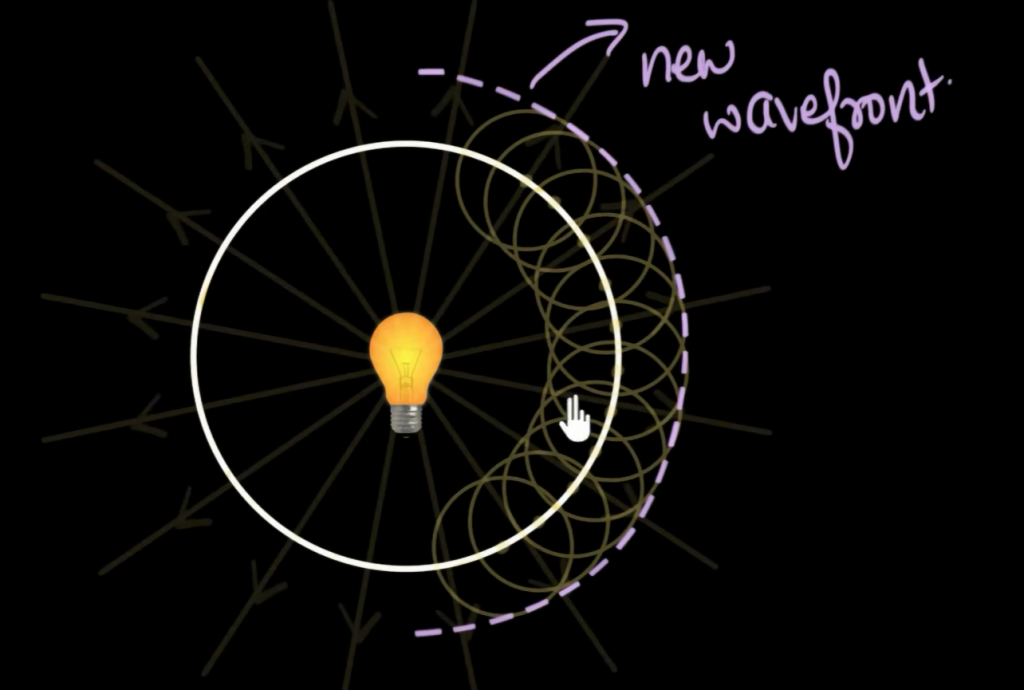
Diffraction effects most noticeable when size of obstacle is ~ to wavelength.
5.2 Diffraction from a Lattice
- X-ray Diffraction (XRD)
X-ray在大多数情况下与lattice发生destructive interference(相消干涉),但在特定角度下可发生constructive interference(相长干涉)
- Systematic absence
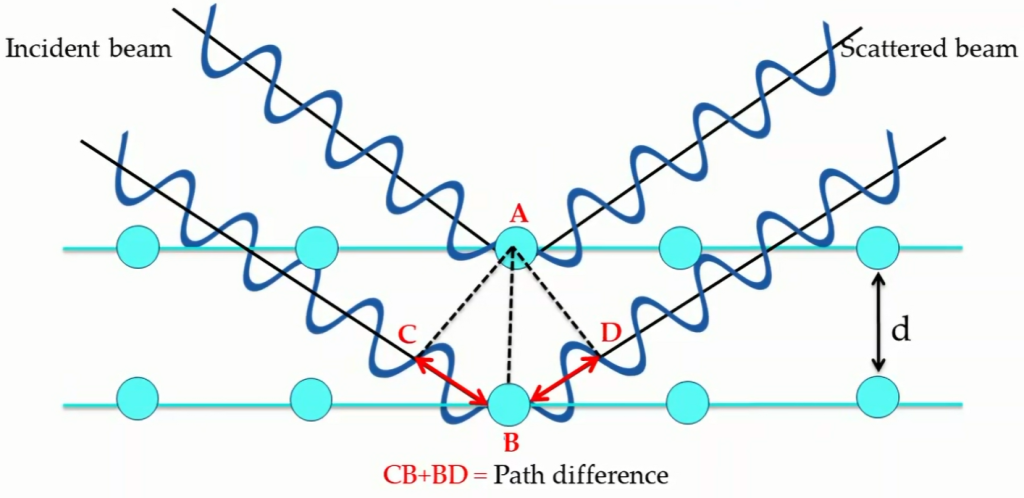
- Bragg’s Law
发生constructive interference的必要条件:
$$n\lambda =2d\sin\theta$$
5.2 Structure Factors
Unit 6: Mechanical Properties
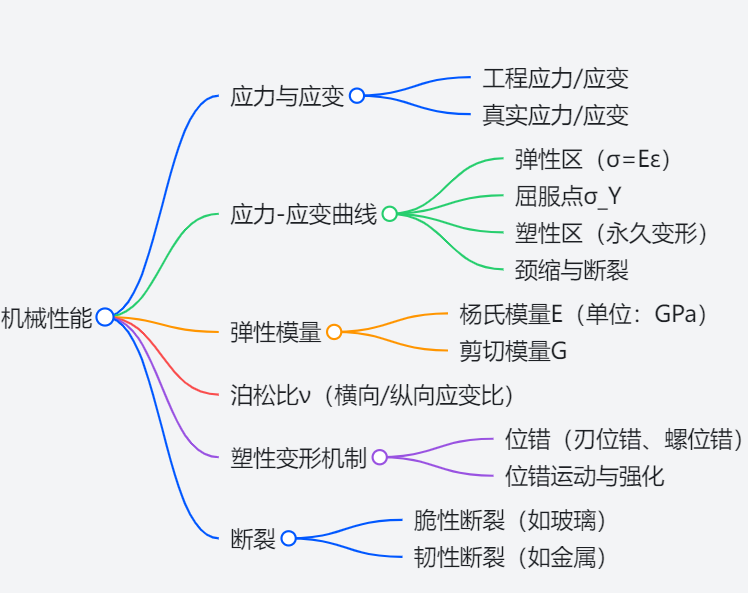
6.1 Stress and Strain
- Stress
$$ \sigma=\frac{F}{A} $$
- Strain
$$ \varepsilon_{true} =\frac{dx}{x} $$
6.2 Stress-Strain Curve
- Elastic Regime
For most of this regime, stress and strain are proportional:
$$ \sigma=E\varepsilon $$
- Young’s Modulus
6.3 Dislocations
Special Relativity
The role of relativity, in physics, is to study how that motion looks from different perspectives.
一个物品在空间中的位置取决于point of reference,即坐标系的选取:
坐标系的移动
从静止的角度看运动,位置是相对的,距离的比率是绝对的.
https://www.youtube.com/watch?v=hTxWAQGgeQw
平移
$$x_{new}=x-\Delta x$$
$$y_{new}=y-\Delta x$$
旋转
$$x_{new}=x\cos\theta-y\sin\theta$$
$$y_{new}=y\cos\theta+x\sin\theta$$
Lorentz Transforms
length contraction
Thermodynamics
Enthalpy
Sublimation
升华过程的焓变是熔化(fusion)和汽化(vaporization)焓变之和:
$$\Delta_{\text{sub}}H^{\ominus} = \Delta_{\text{fus}}H^{\ominus} + \Delta_{\text{vap}}H^{\ominus}$$

Hess’s law
化学反应的总焓变与反应途径无关,只要反应的起始状态(反应物)和最终状态(生成物)相同。这是因为焓($H$)是状态函数,反应的焓变($\Delta_r H$)仅取决于系统终态(产物)与初态(反应物)的焓差,与中间步骤或反应路径无关;热化学方程式可像代数方程一样进行加减、乘以常数等运算。例如,将分步反应的方程式相加(同时焓变相加),消去中间产物,即可得到总反应及其焓变.
一般反应焓变公式
$$\Delta_{r}H_{298}^{\ominus} = \sum \underbrace{\nu_{i}\Delta_{f}H_{298}^{\ominus}(产物)}_{\text{所有产物标准生成焓变之和}} – \sum \underbrace{\nu{i}\Delta_{f}H_{298}^{\ominus}(反应物)}_{\text{所有反应物标准生成焓变之和}}$$
其中 $\nu_{i}$ 是化学计量系数(stoichiometric coefficient),焓是广度性质(extensive quantity),传热与物质的量有关.
一般反应示例
对于 $\alpha A + \beta B \to \gamma C + \delta D$,
$$\Delta_{r}H_{298}^{\ominus} = [\gamma\Delta_{f}H_{298}^{\ominus}(C) + \delta\Delta_{f}H_{298}^{\ominus}(D)] – [\alpha\Delta_{f}H_{298}^{\ominus}(A) + \beta\Delta_{f}H_{298}^{\ominus}(B)]$$
Enthalpy changes of combustion $\Delta_c H^{\ominus}$
- $\Delta_c H^{\ominus}$ 是 1 mol 物质在 1 bar 压力下与氧气完全反应时的焓变,数值通常在 298K 下给出。标注 298K 表示反应物从 298K 开始,产物也在该温度结束;虽然燃烧过程中温度会升高,但关键在于起始和结束温度
- 燃烧焓变总是负值(always negative values),因为燃烧总是放热(gives out heat);计算时必须包含负号,以确保算术运算正确
Enthalpy changes of solution $\Delta_{sol} H^{\ominus}$
1 mol 物质在1 bar 压力下溶于大量溶剂时的焓变
焓变和溶剂量无关
Kirchhoff equation
$$H_{T_2} = H_{T_1} + C_p \Delta T$$
其中,$\Delta T = T_2 – T_1$
对于 1 mol 物质,温度从 $T_1$ 变化到 $T_2$ 时,焓从 $H_{T_1}$ 变为 $H_{T_2}$,变化量由恒压热容 $C_p$ 和温度变化 $\Delta T$ 决定
若已知温度 $T_1 $ 时反应的焓变 $ \Delta H_{T_1} $,则另一温度 $T_2 $ 时的 $ \Delta H_{T_2} $ 可通过反应物和产物的焓随温度变化推导(即Kirchhoff equation):
$$\Delta H_{T_2}^\ominus = \Delta H_{T_1}^\ominus + \Delta C_p (T_2 – T_1) $$
其中 $ \Delta C_p $为反应中产物与反应物热容的差值($ \Delta C_p = C_{p,产物} – C_{p,反应物} $)
Carnot Circle
Heat engine




可逆热机 Reversible engine
可逆热机是可以进行可逆循环的热机,既能从高温热源$T_h$吸热,在低温热源$T_c$放热,完成一个循环;也能逆着该循环,从低温热源$T_c$吸热,在高温热源$T_h$放热;每一个步骤都是可逆的,即过程可无限缓慢(准静态)进行,系统始终处于热力学平衡状态.
$$\eta=1-\frac{T_c}{T_h}$$
在两个给定热源之间工作的任何热机,其效率都不会高于在这两个热源之间工作的可逆热机.

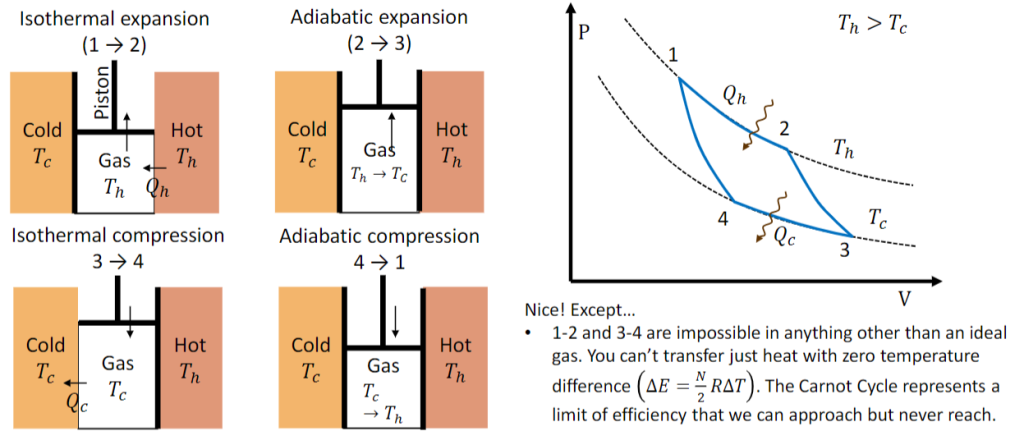
Detailed process
- isothermal expansion
- adiabatic expansion
- isothermal compression
- adiabatic compression


coefficient of performance
$$COP = \frac{T_c}{T_h – T_c}$$
在热机反向运行(如制冷机或热泵)的情境下,COP用于衡量系统的效率,具体物理含义为:系统从低温热源吸收的热量与运行过程中所消耗能量的比值.

COP 定义差异源于对不同系统中 “有用热量” 的界定:制冷系统关注从低温热源提取的 $Q_C$,供热系统关注向高温热源添加的$Q_h$.
Entropy
Entropy is a measure of the microscopic randomness associated with a closed system.
Irreversible process
- General Heat engine efficiency
卡诺循环的最大效率依赖于 “可逆性”;为了更精确地描述过程的可逆程度,需要引入一个新参数 —— 熵,它用于量化过程的可逆性.
由于卡诺循环是可逆循环,在可逆过程中,$\Delta S_{\text{总}}=0$,即系统与外界的总熵变之和为零;因此,高温热源减少的熵等于低温热源增加的熵,即高温热源失去的熵完全转移给了低温热源.
$$\eta’=1-\frac{Q_c’}{Q_h’}$$
- Inequality of efficiencies
$\eta’\leq \eta$,即$1-\frac{Q_c’}{Q_h’}\leq 1-\frac{T_c}{T_h}$
即$\frac{T_c}{T_h}\leq \frac{Q_c’}{Q_h’}\Rightarrow \frac{Q_c’}{T_c’}-\frac{Q_h’}{T_h}\geq0$

Clausius theorem
- For a cycle
$$ \Delta S_{res} = -\oint \frac{dQ}{T} \geq 0 $$
- For a process
$$ \Delta S = \int \frac{1}{T} dQ_{rev} $$
Partial Differentiation
Function of Multiple Variables
total Derivative
当函数所有变量都发生变化,且变量间存在依赖关系,全导数$\frac{dw}{dx}=(\frac{\partial{w}}{\partial{u}})_v\frac{du}{dx}+(\frac{\partial{w}}{\partial{v}})_u\frac{dv}{dx}$
Partial Derivative
- 定义
固定其它变量,仅对其中一个变量求导
- 应用场景
用于分析多元函数中单一变量对结果的影响,如热力学中固定体积$V$,研究内能$U$随温度$T$的变化率$(\frac{\partial{U}}{\partial{T}})_V$
Properties
- symmetry of the second derivatives
$$\left( \frac{\partial}{\partial v} \left( \frac{\partial w}{\partial u} \right)_v \right)_u = \left( \frac{\partial}{\partial u} \left( \frac{\partial w}{\partial v} \right)_u \right)_v$$
Also can be written as:
$$\frac{\partial^2 w}{\partial u \partial v} = \frac{\partial^2 w}{\partial v \partial u}$$
在多元函数的偏导数分析中,函数 $f(x, y)$ 的值可以作为一个变量来处理。
Thermodynamic Potentials, Chemical Potential and Enthalpy
Thermodynamic Potentials
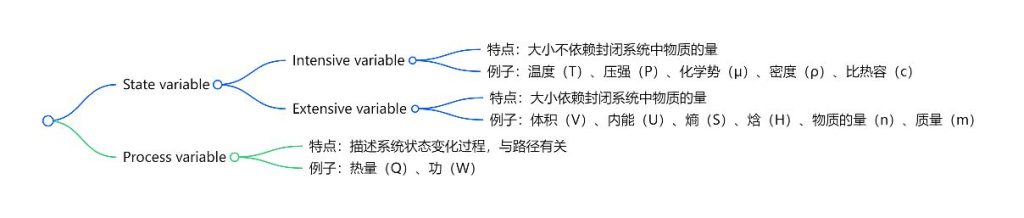
- Intensity variable: the magnitude does not depend on the amount of substance in a closed system.
- Extensive variable: the magnitude depends on the amount of substance present in a closed system
Fundamental Thermodynamic Relation
We have been writing the first law as:
$$\Delta U = Q_{in} + W_{on}$$
But the differential form is of more use (small changes, integrable)
$$dU = dQ_{in} + dW_{on}$$
For a \underline{reversible process}, we have that:
$$dS = \frac{dQ_{rev}}{T} \quad dW_{on} = -P dV$$
And so:
$$dU = T dS – P dV$$
where $ U, S, V $ are state variables! Properties of the material, not the process. This also holds for non – reversible processes.
$$\boxed{dU = T dS – P dV}$$
- chemical potentials
如果存在化学反应,以上公式就遗漏了一个用于解释分子数量变化的项;分子携带能量,因此若不是封闭系统,则必须考虑这部分能量.
$$ dU = TdS – PdV + \sum_{i} \mu_{i} dn_{i} $$
Where $\mu_{i}$ are the $\underline{\text{chemical}\ \text{potentials}}$ of particles of type $i$ and $dn_{i}$ is the number of them. We are literally counting the numbers of molecules and the chemical energy they hold.
$$ dU = TdS – PdV + \sum_{i} \mu_{i} dn_{i} + \sum_{j} X_{j} dx_{j} $$
Thermodynamic Potentials and Free Energy

Thermodynamic Potentials and Maxwell Relations

Relativity

0 条评论