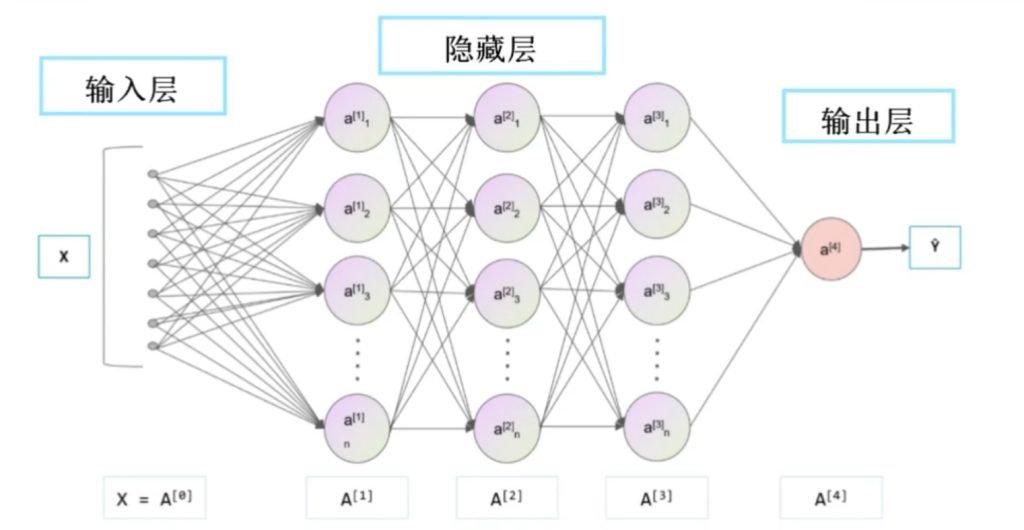
机器学习通过 GNN 模型挖掘数据中的隐含规律,建立电子结构参数与催化性能的映射关系,实现对未计算合金的快速预测,大幅减少计算成本和时间.
摘要
- 催化数据集的构建方法与现有数据集的局限性
- 大模型在催化体系预测中的应用(如吸附能、过渡态计算)
- 通过局域微调算法提升模型精度至化学精度(0.03eV)
- 动态催化模拟的实际案例(如纳米金催化、铜表面重构)
亮点
- 🔍 数据集构建挑战:现有主流数据集(如MP、OC20)多为块体材料,缺乏固液/固气界面数据,团队通过整合金属、二维材料、分子数据库填补了这一空白。
- 🧠 大模型直接预测:训练的大模型可快速计算吸附能、过渡态,但初始误差较大(~0.4 eV),需结合局域微调优化至化学精度(0.03 eV)。
- ⚙️ 局域微调算法:通过少量DFT计算微调特定势能面区域,使模型在未见过体系(如高熵合金)中仍保持高精度,计算速度提升10倍以上。
- 🌡️ 动态催化现象:揭示一氧化碳诱导纳米金颗粒“准液化”及铜表面动态重构,解释了实验观测的低温活性差异(如哈洛塔效应)。
- 🔗 跨学科应用:模型支持催化反应网络构建(如碳一化学)、单原子催化剂设计,并与实验组合作验证理论预测(如原位电镜观测)。
机器学习势函数 #计算催化 #动态模拟 #AIforScience #材料设计
思考
- 局域微调算法是否需要针对不同催化反应类型(如氧化/还原)调整微调策略?
- 如何平衡数据集覆盖广度与计算成本(如高熵合金的多元素组合)?
- 动态催化模拟能否预测实际工业反应条件下的催化剂寿命衰减机制?
视频章节总结
00:00 – ✨ 催化大模型及其应用
该报告主要介绍了基于机器学习势函数构建的催化大模型及其应用。首先介绍了数据集的构建,包括常见吸附物在固体表面的吸附,并利用结构优化和分子动力学模拟来包含平衡态和非平衡态的点。为了提高DFT计算效率,上下表面同时吸附分子,并考虑不同催化反应的中间体和吸附质之间的相互作用。其次,展示了大模型在催化体系中的应用,如结构优化、过渡态计算和分子动力学模拟。通过局域微调算法可以提高大模型的精度,达到化学精度。最后,介绍了神经网络势函数在动态催化中的应用,包括负载催化剂、二氧化钛负载基因体系和铜表面吸附一氧化碳等体系,并结合微观动力学模拟研究了不同cluster的催化反应速率,从而得到动态催化的效应。
00:48 – ✨催化数据集新突破
目前固体表面数据集匮乏,现有数据集多处理快体体系,不适用于催化体系。OC20和OC22是适用于催化体系的数据集,但OC22只包含氧化物,OC20精度较低。研究者希望构建一个包含催化反应所有内容的数据集,供全球科学家使用。他们基于现有数据集,使用DPR和等变图网络等软件进行计算,并用自产数据集微调大模型,效果良好。已将训练好的模型上传至GitHub。微调后能量精度显著提高。即使计算数据集中未出现的复杂体系,也能取得较好结果。数据集构建尽可能包含多种结构,涉及氧化物、金属、气分及界面。研究者展示了从AMD到DPMD的转化过程,并感谢合作者。
03:27 – ✨ 催化ML势函数大模型数据集构建
为了构建用于催化研究的机器学习势函数大模型,研究者们整合了金属、二维材料和分子数据集。为了保证数据的一致性和通用性,所有数据都使用相同的DFT计算参数(例如,cutoff能量为500电子伏特)重新计算。对于分子数据,重新运行了QM9数据集的结构优化和分子动力学模拟,以避免跨程序问题。数据集的构建考虑了原子数和体系的合理分布,以及受力和能量的分布范围。数据提取时,为了平衡数据量,金属和二维材料每五帧取一个结构,分子数据每二十帧取一个结构。未来的工作将致力于提高数据的通用性,例如统一使用合适的交换关联泛函和k点网格密度,并考虑DFT+U的影响。
10:58 – ✨ 机器学习势函数催化大模型:精度与效率提升
该报告讨论了如何构建和应用基于机器学习势函数的催化大模型,以提高计算催化的效率和精度。为了解决不同数据集和计算参数(如赝势、泛函、截断能、加U校正和范德华校正)带来的影响,研究者对数据进行了统一处理。虽然直接使用大模型计算吸附能存在一定误差,但线性标度关系仍然良好。针对过渡态计算,研究者借鉴了OC20N1B的研究,并发现直接使用大模型优化出的过渡态进行单点能量计算仍存在误差。为了达到化学精度(0.034 eV),研究者提出了一种局域微调算法,该算法通过在局部势能面上进行少量DFT计算,然后对大模型进行微调,从而显著提高了计算精度和速度。实验表明,该算法在碳一化学和高熵合金等体系中均表现良好,可在较少的步骤内达到化学精度,并加速结构优化和过渡态计算。
29:57 – ✨ 金纳米颗粒在不同表面的稳定性研究
该研究利用机器学习势函数扩展了原子氧化式负载金纳米颗粒的模拟规模至上万个原子。初期研究对比了氧化硅和氧化式表面负载金纳米颗粒的差异:氧化硅表面疏水,金纳米颗粒易聚集;氧化式表面金纳米颗粒稳定,接触角小。这种稳定性差异源于氧化式中钛的4F轨道可以容纳电子,形成离子相互作用的电荷转移,以及金和氧化式中的氧之间形成较强的金氧共价键。而氧化硅缺乏这些作用,导致金纳米颗粒结合较弱。最后,研究与实验团队合作,通过原位透射电子显微镜观察了金纳米颗粒的聚集过程。
32:01 – ✨纳米金催化与铜表面重构
这段话主要讨论了纳米金催化一氧化碳氧化反应和金属铜上的水煤气变换反应。研究表明,纳米金颗粒的尺寸会影响一氧化碳氧化反应的活性,最佳尺寸约为2纳米。一氧化碳氧化反应的机理可能涉及金纳米颗粒与氧化物载体的界面反应,或者Langmuir-Hinshelwood (LH) 机理。此外,一氧化碳可以诱导金属铜表面的重构,即使初始表面光滑,也会在反应气氛中变得坑洼,形成团簇。神经网络势函数分子动力学模拟可以观察到这些动态过程,表明一氧化碳降低了铜原子的迁移能垒,促进了表面重构。
32:47 – 🔥 金纳米颗粒尺寸与催化活性研究
该研究通过分子动力学模拟,探讨了金纳米颗粒尺寸和一氧化碳吸附对其熔点和催化活性的影响。研究发现,小尺寸金纳米颗粒(小于2纳米)在常温下呈现液态或类液态结构,且一氧化碳吸附显著降低其熔点,并促进金原子的迁移。相比之下,大尺寸金纳米颗粒受一氧化碳吸附的影响较小。这种尺寸依赖性导致了不同的催化反应机理:大颗粒主要通过金-氧化物界面处的MVK机理在高温下反应,而小颗粒则通过金表面的LH机理在低温下反应。这解释了实验中观察到的,小尺寸金纳米团簇催化剂的起始反应温度明显低于大尺寸金纳米颗粒催化剂的现象。研究还利用吉布斯-托马斯方程解释了纳米颗粒尺寸和表面能对熔点的影响,以及一氧化碳吸附如何改变表面能,进而影响熔点。
Reference
0 条评论